本記事ではNGS(次世代シーケンス)実験を勉強中、特にwet研究者の方向けに、NGS解析のおおまかな流れをご紹介します。
NGS(次世代シーケンス)では実験結果を取得するまで、wet作業と呼ばれるシーケンスを行うまでのサンプル処理やシーケンス前処理を行う工程と、dry作業と呼ばれる取得したシーケンス結果を解析し意義あるものにする工程に大きく分かれます。
wet作業は分子生物学に関わる知識、dry作業はコンピュータ処理に関わる知識が求められ、何れかのみに精通して学んできた方が以前は多かったかと思います。ただNGSでは実験結果の取得までに両方の知識が必要となり、特にwetが中心の研究室では、解析がネックとなりNGS実験が出来ないケースも多く見られました。最近では解析フローの自動化や、解析技術やサポートが広まったことにより、実験が出来ないケースは少なくなりましたが、それにより各工程のみを担当し分野外は門外漢で問題なしとなることも多いようです。
もちろんNGS業界全体としては解析に詳しくなくても実験ができるようになったことは喜ばしいのですが、NGS実験のフローをすべて知っていることは、トラブルシューティングの対応など、プラスになってもマイナスになることはありません。また海外などではNGS実験に関わる人間として、解析など多少出来て当たり前と考えている研究室もあるようです。
ここからはNGS解析初心者向けに、シーケンスを行って出力されたデータがどのような処理をされているか、Illuminaシーケンサーの場合のおおまかな流れを紹介します。
BCLファイルをFASTQファイルに変換
シーケンスしたデータは最初、BCLファイルと呼ばれる形式で手にいれることが出来ます。BCLファイルはシーケンスのイメージデータや、クオリティスコア、バーコード情報などが含まれる完全な生データです。BCLファイルは配列がイメージデータのままであり、他の解析ツールで利用できなかったりするため、各種ツールやフォーマットに対応している FASTQファイルに変換します。
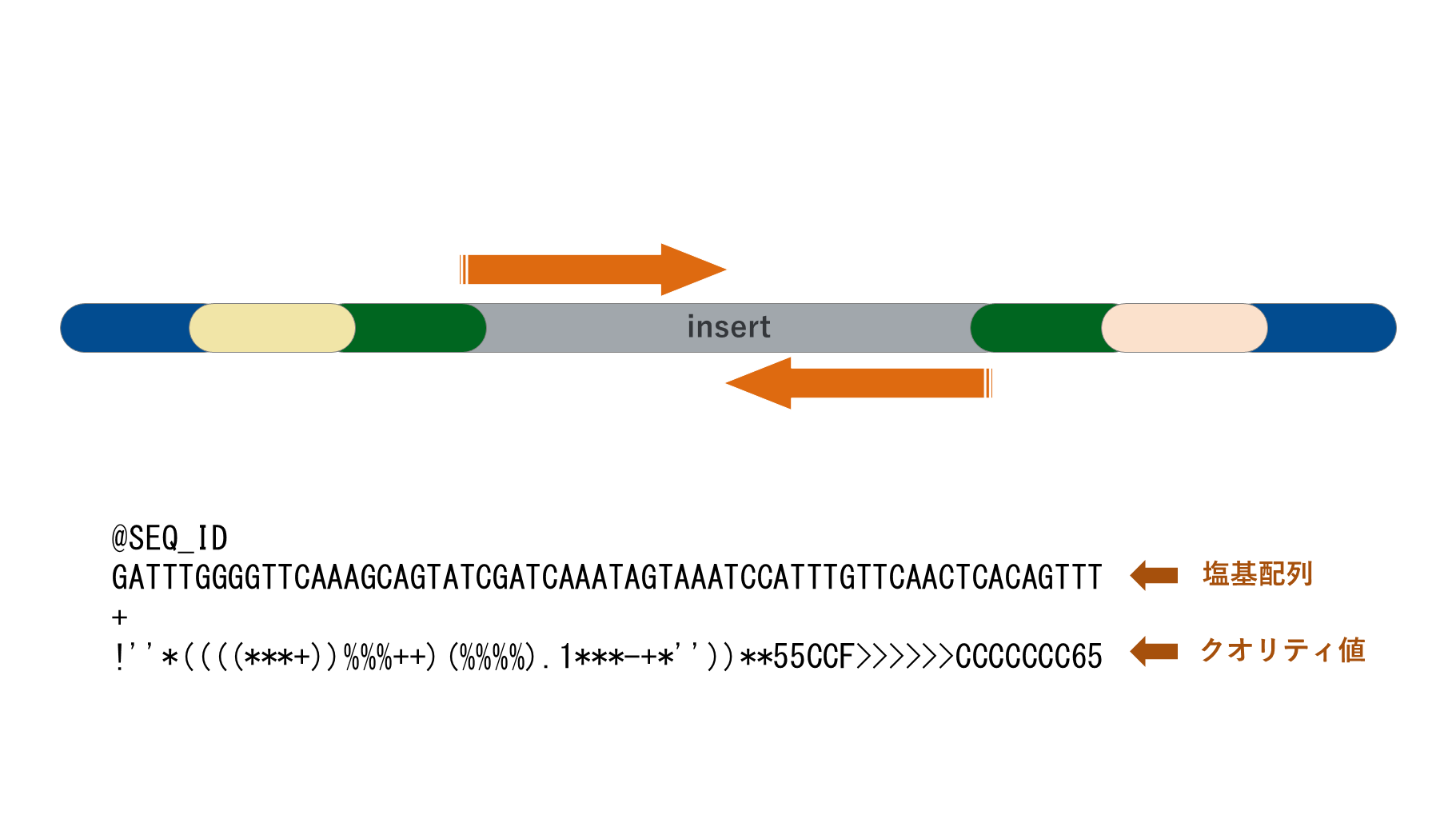
シーケンスした塩基配列とそのクオリティスコアを表記するフォーマット。1行目は”@”で始まり配列のIDと任意でコメントなどを、2行目に実際の塩基配列を、3行目に”+”とIDなどを、4行目に2行目の配列と対応する配列のクオリティ値を記載する。クオリティ値には文字や記号などが表記されているが、ASCII(というもの)を使用しており、1文字に対して2桁の数値で処理できる。

FASTQファイルの前処理
FASTQファイルはすべてのシーケンスデータから作成されていますが、中には不要な配列や信頼性の低いデータが含まれており、この段階で余計なデータを評価、選択していきます。
・クオリティ評価とトリミング
シーケンス結果のクオリティー評価はいろいろな側面があるため、今回の記事では割愛し別記事で紹介する予定です。解析作業としては、クオリティが低いと思われるリードを除いたり、リード中の両末端などクオリティの低い配列を取り除きます。
・アダプタートリミング
作製したライブラリーのインサート長とリード長次第では、読んだ配列中にアダプター配列が含まれることがあります。サンプルとして読んだはずの配列にアダプター配列が含まれていると、その後の解析や実験結果に影響があるので、アダプター配列を除去したりマスクしていく処理が必要になります。

目的にあわせたデータ解析を進める
ここからの解析は目的とする実験と結果によって工程が違ってきます。下記は代表的な実験例です。
3-1. 配列をつなぎあわせて新規配列を得る
参照配列がない生物のゲノムを決定する実験などで進める工程です。得られたリード配列の共通分を繋げて、生物のは配列を決定していきます。繋ぎあわせる工程はアセンブリ、繋ぎあわせた配列はスキャフォールドと呼ばれます。
3-2. 参照配列と比較する
健常者とある疾患のサンプルを比較する実験などで進める工程です。ヒトなどすでに参照配列が決定している場合、サンプルで取得したシーケンス配列を参照配列とマッチする配列と比較して決定していきます。その中で疾患サンプルのみ特定のSNP変異が多いといった違いを見つけていきます。シーケンス結果を参照配列とマッチしていくことをマッピング、変異を見つけることはコールと呼ばれます
3-3. 各リードの数を比較して遺伝子発現量を比較する
RNA-Seqなど遺伝子発現量を解析する実験で進める工程です。取得した配列を参照配列にマッピングすることは3-2と変わりませんが、発現量の比較ではマッピングしたリード数を比較して、解析を進めていきます。
実際の解析では、クオリティ評価のカットオフ値をどうするか、遺伝子発現量のリード数測定は長さで補正する必要があるなど、追加の工程が存在しています。詳細に関しては、今後の記事で少しずつ紹介していく予定です。